Automating large numbers of tasks¶
Researchers often need to perform a number of calculations that vary only in their initial conditions or input parameters. Such tasks naturally arise when exploring the predictions of a model over a range of parameters or when testing a numerical calculation for convergence. Automating these tasks is critical, both for computational efficiency and to minimize human error and ensure the calculation is reproducible.
This tutorial will discuss a number of techniques and tools available on Midway for automating the running of tasks and their respective advantages and disadvantages. In particular we will focus on those tasks which are entirely independent of each other and where the total number of tasks is known (and fixed).
When deciding between the various techniques, users should consider the following:
- overhead or cost of starting each task (including cost of starting remote processes)
- waiting time each batch job will spend in the queue
- total number of jobs compared to available resources and limits
- the average calculation time per job and its variance
- the cost in developer time needed for a given solution
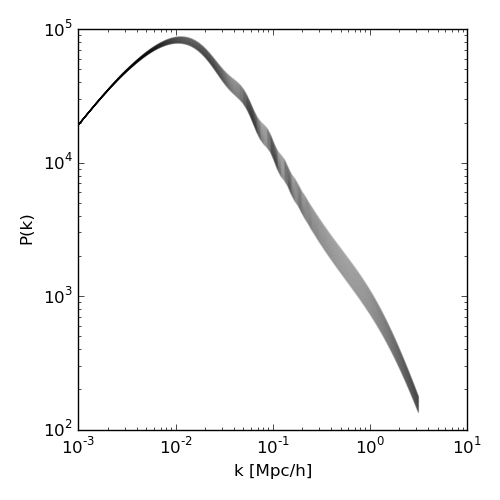
As a semi-realistic example, this tutorial will use the CLASS code to compute the non-linear matter power specturm for a range of the neutrino parameter N_eff.
CLASS and classy¶
CLASS [1104.2932] is a Boltzmann code similar to CMBFAST, CAMB, and others. It can be used to compute a number of large-scale structure and CMB observables and since it is designed to perform calculations within MCMC-type likelihood analyses, it is a good option for these exercises.
To install CLASS and its Python wrapper, classy, use the following commands:
module load git python/2.7-2014q3
git clone https://github.com/lesgourg/class_public
cd class_public
make
Note
This will install the Class python wrappers in your .local directory while remembering where you compiled CLASS. Make sure you delete them both after completing the exercises.
classy is a Python wrapper for the CLASS code which we will use for convenience. It allows us to avoid the common issue of dealing with parameter files
The code that uses classy to compute the non-linear matter power spectrum for a given N_eff is as follows:
#!/bin/env python
def compute_pk(N_eff=3.04, output="pk.dat"):
import numpy as np
from classy import Class
# initialize Class with default parameters save N_eff
c = Class()
c.set({'output':'mPk', 'non linear':'halofit', 'N_eff':N_eff})
c.compute()
with open(output, "w") as output:
for k in np.logspace(-3., 0.5, 100):
print >>output, "%.6e %.6e" % (k, c.pk(k, z=0.0))
c.struct_cleanup()
# if necesssary for visualization, add a 5 second delay
#import time; time.sleep(5)
# perform work associated with i out of N steps
def work(i, N=100):
N_eff_min = 2.5
N_eff_max = 4.5
N_eff = (N_eff_max-N_eff_min)*float(i)/float(N) + N_eff_min
output = "pk_{}.dat".format(i)
compute_pk(N_eff, output)
# collect the resulting matter power spectra and plot
def plot_pk(file_pattern="pk_*.dat",output="pk.png"):
import pylab, glob
import numpy as np
f = pylab.figure(figsize=(5,5))
for data in glob.glob(file_pattern):
(k, pk) = np.loadtxt(data, unpack=True)
f.gca().loglog(k, pk, color='k', alpha=0.05)
f.gca().set_xlabel("k [Mpc/h]")
f.gca().set_ylabel("P(k)")
f.tight_layout()
f.savefig(output)
if __name__ == "__main__":
import sys
if len(sys.argv) == 3:
work(int(sys.argv[1]), int(sys.argv[2]))
else:
plot_pk()
print "done"
Run this code without any input parameters to generate the following png figure from the resulting output files.
Parallel Programming¶
Implementing your own parallel scheme is useful when the tasks are not completely independent, or represent only a part of a larger, non-trivial workflow. Calculations that rely on large pre-calculated tables or have costly initilization may also benefit from an explicit scheme, as those costs can be amortized over a larger number of tasks.
Multiprocessing¶
Smaller numbers of tasks can be divided amongst workers on a single node. In high level languages like Python, or in lower level languages using threading language constructs such as OpenMP, this can be accomplished with little more effort than a serial loop. This example also demonstrates using Python as the script interpreter for a Slurm batch script, however note that since Slurm copies and executes batch scripts from a private directory, it is necessary to manually add the runtime directory to the Python search path.
#!/bin/env python
#SBATCH --job-name=multiprocess
#SBATCH --output=logs/multiprocess_%j.out
#SBATCH --time=01:00:00
#SBATCH --partition=kicp
#SBATCH --account=kicp
#SBATCH --nodes=1
#SBATCH --exclusive
import multiprocessing
import sys
import os
# necessary to add cwd to path when script run
# by slurm (since it executes a copy)
sys.path.append(os.getcwd())
from pk import work
# get number of cpus available to job
try:
ncpus = int(os.environ["SLURM_JOB_CPUS_PER_NODE"])
except KeyError:
ncpus = multiprocessing.cpu_count()
# create pool of ncpus workers
p = multiprocessing.Pool(ncpus)
# apply work function in parallel
p.map(work, range(100))
MPI¶
Process and threaded level parallelism is limited to a single machine. To
#!/bin/env python
#SBATCH --job-name=mpi
#SBATCH --output=logs/mpi_%j.out
#SBATCH --time=01:00:00
#SBATCH --partition=kicp
#SBATCH --ntasks=100
from mpi4py import MPI
from pk import work
comm = MPI.COMM_WORLD
rank = comm.Get_rank()
size = comm.Get_size()
work(rank, size)
MPI programs and Python scripts must be launched using mpirun as shown in this Slurm batch script:
#!/bin/bash
#SBATCH --job-name=mpi
#SBATCH --output=logs/mpi_%j.out
#SBATCH --time=01:00:00
#SBATCH --partition=kicp
#SBATCH --account=kicp
#SBATCH --ntasks=100
module load mpi4py/1.3-2014q3+intelmpi-4.0
mpirun python mpi_pk.py
In this case we are only using MPI as a mechanism to remotely launch tasks on distributed nodes. All processes must start and end at the same time, which can lead to waste of resources if some job steps take longer than others.
Slurm task per job¶
In some cases it may be easiest to wrap the job submission in a script or bash command, taking advantage of the fact that Slurm will pass on environment variables defined at the time of job submission (this is also why you can load modules before submitting a job rather than inside the job script itself).
for i in {0..10}; do export i; sbatch job.sbatch; done
#!/bin/bash
#SBATCH --job-name=job
#SBATCH --output=logs/job_%j.out
#SBATCH --time=01:00:00
#SBATCH --partition=kicp
#SBATCH --account=kicp
#SBATCH --ntasks=1
python pk.py $i 100
Note
It can be difficult to monitor and jobs submitted in this way. Be sure to use a unique
job-name so you can identify a particular job group with commands like squeue and scancel,
e.g. scancel -n class
GNU Parallel¶
GNU Parallel is a tool for executing tasks in parallel, typically on a single machine. When
coupled with the Slurm command srun, parallel becomes a powerful way
of distributing a set of tasks amongst a number of workers. This is particularly useful when
the number of tasks is significantly larger than the number of available workers (Slurm’s
--ntasks).
#!/bin/sh
#SBATCH --time=01:00:00
#SBATCH --output=logs/parallel.log
#SBATCH --partition=kicp
#SBATCH --account=kicp
#SBATCH --ntasks=32
#SBATCH --exclusive
module load parallel
# the --exclusive to srun makes srun use distinct CPUs for each job step
# -N1 -n1 allocates a single core to each task
srun="srun --exclusive -N1 -n1"
# --delay .2 prevents overloading the controlling node
# -j is the number of tasks parallel runs so we set it to $SLURM_NTASKS
# --joblog makes parallel create a log of tasks that it has already run
# --resume makes parallel use the joblog to resume from where it has left off
# the combination of --joblog and --resume allow jobs to be resubmitted if
# necessary and continue from where they left off
parallel="parallel --delay .2 -j $SLURM_NTASKS --joblog logs/runtask.log --resume"
# this runs the parallel command we want
# in this case, we are running a script named runtask
# parallel uses ::: to separate options. Here {0..99} is a shell expansion
# so parallel will run the command passing the numbers 0 through 99
# via argument {1}
$parallel "$srun python pk.py {1} 100 > logs/parallel_{1}.log" ::: {0..99}
This job is submitted as with any Slurm batch job
$ sbatch parallel.sbatch
and will appear as a single job in the queue, assigned as many nodes/cores as was requested:
$ squeue -u $USER -p kicp
JOBID PARTITION NAME USER ST TIME NODES NODELIST(REASON)
11511073 kicp parallel drudd R 0:00 2 midway[203,390]
GNU Parallel maintains a log of the work that has already been done, along with the exit value of each step (useful for determining any failed steps).
$ head logs/runtask.log
Seq Host Starttime Runtime Send Receive Exitval Signal Command
1 : 1422211597.529 6.467 0 0 0 0 srun --exclusive -N1 -n1 python pk.py 0 100 > logs/runtask.0
2 : 1422211597.732 6.466 0 0 0 0 srun --exclusive -N1 -n1 python pk.py 1 100 > logs/runtask.1
3 : 1422211597.935 6.466 0 0 0 0 srun --exclusive -N1 -n1 python pk.py 2 100 > logs/runtask.2
4 : 1422211598.138 6.464 0 0 0 0 srun --exclusive -N1 -n1 python pk.py 3 100 > logs/runtask.3
In our case we requested the output from each work step be directed to a file logs/runtask.{1},
allowing us to peform further diagnostics if necessary. The parallel option --resume creates a file
parallel.log which allows GNU Parallel to resume a job that has been stopped due to failure or by hitting
a walltime limit before all tasks have been completed. If you need to rerun a GNU Parallel job, be sure
to delete parallel.log or it will think it has already finished!
Note
More information may found in the RCC documentation section Parallel Batch Jobs.
Slurm Job array¶
Most HPC job schedulers support a special class of batch job known as array jobs (or job arrays). Slurm support for job arrays is relatively new and is undergoing active development. The GNU Parallel solution in the previous section was developed at RCC because of the former lack of Slurm array jobs.
An example Slurm array job submission script is as follows:
#!/bin/bash
#SBATCH --job-name=array
#SBATCH --output=logs/array_%A_%a.out
#SBATCH --array=0-99
#SBATCH --time=01:00:00
#SBATCH --partition=kicp
#SBATCH --account=kicp
#SBATCH --ntasks=1
# the environment variable SLURM_ARRAY_TASK_ID contains
# the index corresponding to the current job step
python pk.py $SLURM_ARRAY_TASK_ID 100
This looks very similar to our single-job batch submission script, but with the addition
of the --array feature. In this case, the --array=0-99 will cause 100
individual array-tasks to be created when this job script is submitted. Each array task
is a copy of the master script, with an environment variable called SLURM_ARRAY_TASK_ID
set to the index of the array task. As with the GNU Parallel example, we simply pass the
value of that environment variable into our program to perform that piece of work.
Each array job will enter the queue and run when resources are available for it, as if we had submitted each job manually (this is merely a highly-convenient shortcut).
Array job indices do not need to be a linear range, and can be specified in a number of ways. For example:
A job array with index values of 1, 2, 5, 19, 27:
#SBATCH --array=1,2,5,19,27
A job array with index values between 1 and 7 with a step size of 2 (i.e. 1, 3, 5, 7):
#SBATCH --array=1-7:2
The %A_%a construct in the output and error file names is used to generate unique output and error files
based on the master job ID (%A) and the array-tasks ID (%a). In this fashion, each array-tasks will be able to
write to its own output and error file.
When we submit a job array, we will see the master process, as well as any submitted, running or otherwise
not completed array tasks with the naming convention %A_%a.
$ squeue
JOBID PARTITION NAME USER ST TIME NODES NODELIST(REASON)
11510950_[10-99] kicp array drudd PD 0:00 1 (None)
11510950_0 kicp array drudd R 0:00 1 midway185
11510950_1 kicp array drudd R 0:00 1 midway185
11510950_2 kicp array drudd R 0:00 1 midway185
11510950_3 kicp array drudd R 0:00 1 midway185
11510950_4 kicp array drudd R 0:00 1 midway185
11510950_5 kicp array drudd R 0:00 1 midway185
11510950_6 kicp array drudd R 0:00 1 midway185
11510950_7 kicp array drudd R 0:00 1 midway185
11510950_8 kicp array drudd R 0:00 1 midway185
11510950_9 kicp array drudd R 0:00 1 midway185
We can cancel all array tasks associated with the master job ID as before, or cancel any subset of them using
the naming convention %A_%a
# cancel job steps 10-100
$ scancel 11510950_[10-100]
# cancel all remaining job steps
$ scancel 11510950
What happens if we try to submit a job with, say, --array=0-1000?
sbatch: error: Batch job submission failed: Job violates accounting/QOS policy (job submit limit, user's size and/or time limits)
The number of jobs a user may have submitted (not running!) at any given time is limited, in order to avoid overloading the scheduler and to ensure users cannot accidentally swamp the machines with malformed job submissions. To see how many jobs can be run or submitted for a given partition+QOS, use the following command:
$ sacctmgr list qos kicp format=maxjobsperuser, maxsubmitjobsperuser
MaxJobsPU MaxSubmitPU
--------- -----------
64 128
The ability to submit array jobs which are larger than MaxSubmitPU is coming in a newer
version of Slurm. Until then, each job should handle more than one piece of work, or the GNU Parallel
solution should be used. For more information, see the RCC documentation section Job Arrays.
Note
The current version of Slurm on Midway (14.03) is the first to support Slurm job arrays, and does not support all features described by the current docuemntation. RCC plans to upgrade Slurm to 14.11 in the near future which will allow array jobs to control the number of active job submissions.
Workflow Tools¶
Other specialized tools have been created for handling parallel workflows, particularly in specific domains or for specific analysis or computational codes. Developing such tools is an open area of research.
Swift¶
Swift is a scripting language created at the University of Chicago Computation Institute designed to support portable, parallel workflows. It will handle transferring files to and from various computing platforms, and allows for complex dependencies. Because of this, however, it requires very specific definitions of both the application to be run and the platform (site, in Swift terminology).
For more information see the Swift Midway tutorial.